背景介紹
電催化水分解作為一種可持續氫氣生產的前景技術,受到了廣泛關注。然而,陽極的氧氣析出反應(OER)存在反應動力學緩慢的問題,極大地限制了水分解的效率。因此,深入理解OER催化機制,對于設計高效OER催化劑具有重要意義。根據OER過程中關鍵反應中間體的轉化步驟,OER機制中涉及兩種主流反應路徑:吸附物演變機制(AEM)和晶格氧機制(LOM)。在AEM中,金屬位點作為氧化還原中心,涉及多個高度相關的氧中間體,導致其最低過電位的熱力學限制為0.37 V。對于LOM,氧位點被激活為氧化還原中心,參與氧的形成,從而突破了AEM的比例關系限制,具有更有利的熱力學。然而,主導的LOM路徑由于涉及晶格氧以及氧空位的形成和填補,容易導致催化劑的結構不穩定和催化性能下降。毫無疑問,通過同時激活金屬和晶格氧位點的氧化還原反應構建AEM-LOM兼容的機制,可以結合每條路徑的優點,并調和OER的催化活性和穩定性。然而,在當前以單一組分和配位環境為特征的催化系統中,電子轉移過程要么發生在金屬位點,要么發生在晶格氧位點,這取決于金屬和氧帶相對于費米能級的位置。因此,開發一個能夠同時激活金屬和晶格氧氧化還原對的耦合催化系統仍面臨重大挑戰。
過渡金屬羥基氧化物(MOOH,M = Fe、Co或Ni)是在氧氣析出反應(OER)電氧化條件下不可逆結構重構的產物,普遍被認為是真正的活性物質,其中基于NiFe的羥基氧化物正逐漸取代商業化的RuO2和IrO2,成為OER催化劑的基準。特別是,一些近期報告發現,OER重構衍生的高價金屬中心的羥基氧化物具有觸發晶格氧機制(LOM)以提高OER活性的潛力,這為通過電催化重構策略構建具有合適OER路徑的催化劑提供了重要思路。然而,催化劑的電催化結構重構通常僅限于近表面納米尺度,導致活性組分的利用率較低。NiMoO4水合物具有由四個邊共享的NiO6八面體與MoO4四面體連接而成的三維網絡,并通過陽極電位驅動的結晶水和Mo的共同浸出展現出良好的結構自重構特性,因此可以被選作適合的自犧牲前催化劑,以構建具有靈活配位結構的氧羥化物。
本文要點
1. 在這項研究中,NiMoO4水合物被用作預催化劑,同時通過化學蝕刻共同引入Fe和S作為調節劑,誘導產生豐富的結構缺陷,并促進在電化學激活過程中完全重構為R-NiFeOOH@SO4活性物質。
2. 表征證據表明,陽極激活觸發了金屬和晶格氧位點的氧化還原過程,并涉及氧空位的形成和填補。
3. 此外,通過原位衰減全反射傅里葉變換紅外光譜(ATR FTIR)和18O同位素標記差分電化學質譜(DEMS),可以確認AEM-LOM兼容的OER催化機制,其中R-NiFeOOH@SO4在Fe和S物種的共同調節下,賦予了同時優化的AEM和LOM路徑。
4.密度泛函理論(DFT)計算表明,引入的Fe作為AEM路徑的活性位點優化了OER中間體的吸附,而引入的S顯著刺激了晶格氧的活性,并增加了OER的LOM路徑占有率。在Fe和S的共同調節下,R-NiFeOOH@SO4系統實現了AEM和LOM路徑的協同催化,最大限度地利用了表面金屬和氧活性位點,提升了OER的催化活性。
圖文介紹

圖1.催化劑的制備。a,納米珊瑚狀 NiMoO4.xH2O@Fe,S 的合成路線,插圖顯示了納米棒表面結構的演變。圖 b、f 為NiMoO4.xH2O,c、g 為 NiMoO4.xH2O@Fe,S,d、e、h 為 R-NiFeOOH@SO4 的 FESEM、TEM 和高分辨率 TEM 圖像。圖 i 為 R-NiFeOOH@SO4 的 HAADF-STEM 圖像及相應的 Ni、Mo、O、Fe、S 的 EDS 元素分布。

圖2. 催化劑電子和配位結構表征。高分辨率 XPS 結果顯示了 (a) Ni 2p、(b) O 1s、(c) Fe 2p 和 (d) S 2p 的峰值,比較了獲得的樣品在 CV 活化前后的變化。(e) 是獲得樣品和選定參考材料的歸一化 Ni K 邊和 (f) Fe K 邊 XANES 光譜。(g) 是獲得樣品在 CV 活化前后 Ni K 邊光譜的傅里葉變換 k3χ(R) 圖。
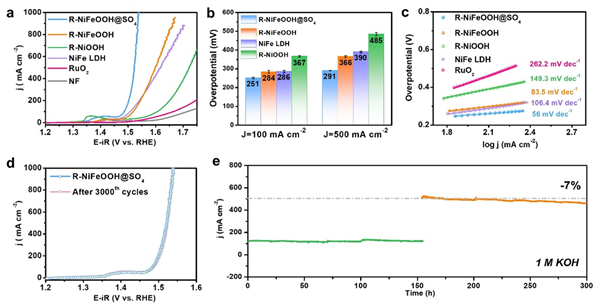
圖3. 催化劑OER性能表征。(a) OER 極化曲線,進行了 80% 的 iR 補償(電極工作面積為 0.5 cm × 0.5 cm,補償所用的溶液電阻約為 2.3 ± 0.2 Ω);(b) 催化劑在 100 和 500 mA cm-2 下的相應過電位;(c) 催化劑的 Tafel 圖;(d) 3000 次 CV 循環前后的 LSV 曲線;(e) R-NiFeOOH@SO4 在 1 M KOH 中于 0.65 和 0.85 V(vs Hg/HgO)下的無 iR 補償的計時安培曲線。
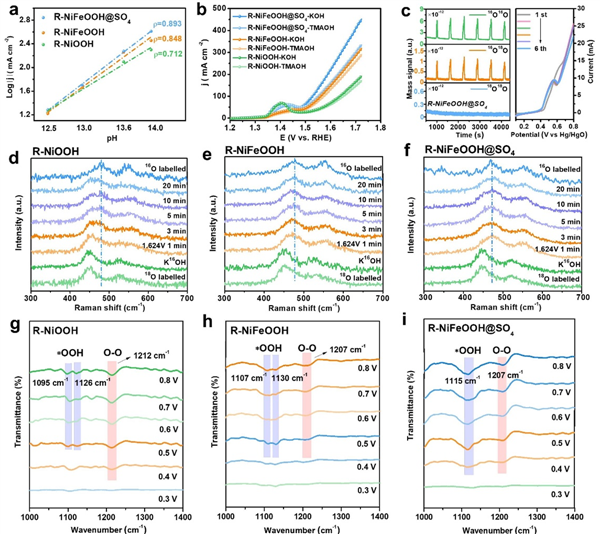
圖 4:AEM-LOM OER 催化分析。 (a) 催化劑在相對于 RHE 的 1.7 V 電位下的電流密度對數與 pH 的關系。 (b) 催化劑在 1 M KOH 和 1 M TMAOH 中的 OER 極化曲線,均未進行 iR 補償。 (c) R-NiFeOOH@SO4 的 DEMS 信號相對于時間的檢測結果,未對 16O16O、16O18O 和 18O18O 進行任何校正或減法處理,及相應的 LSV 曲線,未進行 iR 補償。(d) 18O 標記的 R-NiOOH、(e) R-NiFeOOH 和 (f) R-NiFeOOH@SO4 在 0.1 M KOH 中于 1.624 V 相對于 RHE 測得的原位拉曼光譜。(g) R-NiOOH、(h) R-NiFeOOH 和 (i) R-NiFeOOH@SO4 的原位 ATR-FTIR 光譜。

圖5. DFT理論計算。(a) 態密度, (b) 示意能帶圖(UHB 為上 Hubbard 帶,LHB 為下 Hubbard 帶), (c) Ni-O 鍵的晶體軌道COHP。 (d) Ni 位點和 (e) Fe 位點的 AEM 途徑的 Gibbs 自由能圖,以及 (f) LOM 途徑的 Gibbs 自由能圖。(g) AEM 和 LOM 反應的速率決定步驟(RDS)的能量障礙。
本文信息
Luo, X., Zhao, H., Tan, X. et al. Fe-S dually modulated adsorbate evolution and lattice oxygen compatible mechanism for water oxidation. Nat. Commun. 15, 8293 (2024).
DOI: 10.1038/s41467-024-52682-y
https://www.nature.com/articles/s41467-024-52682-y