原創 景行 新威 2022-09-10 11:55 發表于廣東
01 導讀
為了滿足水電解工業制氫的需求,OER催化劑需要滿足大電流密度(≥1 Acm2)、低過電位(<300 mV)和長期耐久性(超過1000 h)的要求。然而,到目前為止,為了達到1 A cm2及以上的大電流密度,其過電位仍然太大,無法實際應用于水電解的大規模制氫。
02 成果背景
近日,武漢理工大學木士春和南京曉莊學院劉蘇莉等提出了一種“打破對稱性”的策略,通過用Ru原子部分取代鐵鈷層狀雙氫氧化物(FeCo-LDH)來促進FeCo-LDH的電子轉移。這有效地調整了催化劑的界面相互作用和配位/電子環境,極大地提高了OER/HER的催化活性,以及在大電流密度不飽和配位下的整體水分解反應。相關工作以“Breaking the symmetry of single-atom catalysts enables an extremely low energy barrier and high stability for large-current-density water splitting”為題發表在Energy & Environmental Science上。
03 關鍵創新
設計的催化劑對于水分解反應只需要1.52 V就能達到1000 mA cm2的電流密度,而在1000 h后幾乎沒有變化。
04 核心內容解讀
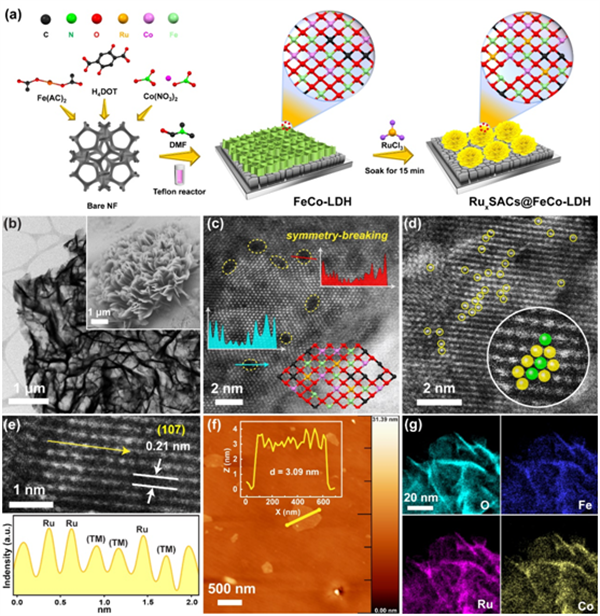
圖1 (a)合成圖示意圖,(b)TEM和FE-SEM圖像,Ru1 SACs@FeCo-LDH的HAADF-STEM圖像,(c) 黃色虛線圈出的缺陷,(d)黃色圓圈標記的亮點代表單個Ru原子。(e) 圖像中白線之間的窄線(TM=Fe,Co)。(f)AFM圖像和相應的AFM高度剖面(內部插圖),(g) Ru1 SACs@FeCo-LDH的元素能譜圖。@ The Authors
Rux SACs@FeCo-LDH采用混合溶劑策略構建。如圖1(a)所示,Rux SACs@FeCoLDH是由首先使用Ni泡沫(NF)作為襯底生長FeCo-LDH陣列,然后在FeCo-LDH陣列上原位生長Ru單原子位點形成的。這打破了原來的對稱性,有利于催化反應。由于催化劑的原位生長,Fe、Co、Ru的含量極低,使得X射線衍射難以觀察到典型的峰。
TEM和場發射掃描電鏡(FE-SEM)圖像顯示,Ru1 SACs@FeCoLDH仍然保持了均勻的三維多孔結構,由相互連接的超薄納米片組成,表面光滑,邊緣尖銳(圖1(b)所示)。通過將實驗球差校正后的HAADF-STEM圖像與仿真結果進行比較,識別出不同的堆疊序列,如圖1(c)所示。放大后的HAADF-STEM圖像顯示了催化劑詳細的界面對稱破缺結構,說明在LDH表面形成了多原子界面,增加了活性表面積和活性位點。同時,觀察到的亮點(圖1(d)中用黃色圓圈表示)對應于單個Ru原子。此外,作者還進行了線性掃描分析,進一步證明了Ru原子的孤立狀態(圖1(e)),但由于Fe或Co原子的原子序數相近,無法區分。圖1(f)顯示了Ru1 SACs@FeCo-LDH納米片的超薄性質,其厚度僅為3.09 nm。能量色散X射線(EDX)光譜進一步證明了Fe、Co和Ru的均勻分布(圖1(g))。
此外,利用Co、Fe和Ru k邊X射線吸收近邊結構(XANES)和擴展X射線吸收精細結構(EXAFS)光譜進一步研究了Ru1 SACs@FeCo-LDH的配位環境和化學狀態。Ru1 SACs@FeCoLDH和FeCo-LDH樣品的Fe k邊XANES譜在大約7133.2 eV處有明顯的前邊峰(圖2(a)),這些樣品的吸收邊緣與三氧化二鐵的吸收邊緣很接近,表明原子分散的Fe物種攜帶+3的正電荷。同時,與FeCo-LDH相比,Ru1 SACs@FeCo-LDH的Fe k邊向較低的光子能量略有轉變,證實了Ru-O-Fe結構的形成。此外,與FeCo-LDH相比,Ru1 SACs@FeCo-LDH的Co k邊的吸收邊緣也表現出相同的輕微的負偏移,位于CoO和Co3O4之間,表明Co物種的價態在+2和+3之間(圖2(b))。這些基于XANES分析結果的觀察結果與XPS分析的結果一致,表明Ru周圍的電子可以轉移到Fe和Co。圖2(c)顯示的Ru1 SACs@FeCo-LDH和Ru2 SACs@FeCo-LDH的Ru k邊XANES譜顯示邊緣能量在Ru箔和RuO2之間,表明Ru的氧化態在0和+4之間,證實了Ru原子的部分未占據的3d軌道帶正電荷。
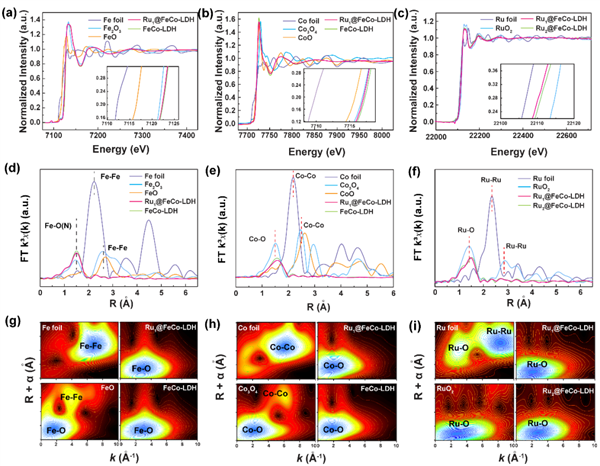
圖2 (a)分別以Co箔和標準CoO或Co3O4粉末為參考,得到的Ru SACs@FeCo-LDH的(a)Fe K邊、(b)Co K邊和(c)Ru K邊XANES譜。(d)在Fe箔、三氧化二鐵、氧化亞鐵、Ru1 SACs@FeCo-LDH和FeCo-LDH的R空間上的傅里葉變換(FT)。(e)Co箔、Co3O4、CoO、Ru1 SACs@FeCo-LDH和FeCo-LDH的R空間上的FT。(f)Ru箔、RuO2和Rux SACs@FeCo-LDH的R空間上的FT。(g-i)分別為(b)Co K邊、Fe K邊和Ru K邊的EXAFS光譜的FT k3加權函數。@ The Authors
如圖2(d)所示,Fe元素的EXAFS光譜(傅里葉變換,無相位校正)在R=1.5 ?處有一個峰值,對應于Ru1 SACs@FeCo-LDH、三氧化二鐵和氧化亞鐵的Fe-O散射。可以看出,在2.1和2.5 ?左右有兩個顯著的配位峰,分別對應于鐵箔、三氧化二鐵和氧化亞鐵的Fe-Fe散射。Ru1 SACs@FeCo-LDH的Co k邊的EXAFS譜中典型的峰出現在1.6 ? (圖2(e)),這是由于LDH上的原子Co與O配位。
圖2(f)中的FTR空間譜清楚地表明,Rux SACs@FeCo-LDH沒有Ru-Ru鍵的散射峰,而有Ru箔的主峰,表明Rux SACs@FeCo-LDH中不存在Ru-Ru鍵,這反映了Ru元素在Ru1 SACs@FeCo-LDH和Ru2 SACs@FeCo-LDH中的單原子色散。
為了更深入地了解原子的配位情況,作者利用小波變換(WT-EXAFS)對Fe、Co和Ru的EXAFS結果進行了變換,該變換可以同時提供關于R空間和K空間的信息。如圖2(g)-(i)所示,Rux SACs@FeCo-LDH僅在2.4 ?處出現了Fe(Co,Ru)-O鍵的特征峰,這可以歸類為原子Fe(Co,Ru)-O物種。Fe-Fe、Co-Co和Ru-Ru配位的缺失消除了Ru團簇的存在。此外,Ru-O鍵具有單原子特征,這也證實了單Ru原子的存在,因此Rux SACs@FeCo-LDH實際上是FeCo-LDH負載和穩定的單原子Ru催化劑。
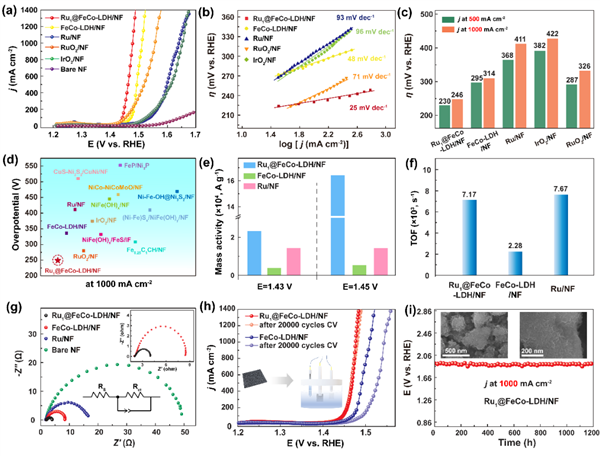
圖3在1.0 M氫氧化鉀中的(a)極化曲線;(b)為相應的Tafel圖;OER電催化在500 mA cm2和1 A cm2處的過電位的(c)比較;(d)在1.0 M氫氧化鉀溶液中,與最近報道的OER催化劑在1 A cm2處的過電位進行比較;(e)不同過電位下MA的比較;(f)電位依賴的TOF曲線;(g)Nyquist圖;(h)Ru1 SACs@FeCo-LDH和FeCo-LDH在20 000個CV循環前后的極化曲線;(i)Ru1 SACs@FeCo-LDH在維持1 A cm2持續1200 h所需電位的時間演化下的時間-電位測量曲線。@The Authors
為了探討Ru單原子改性的FeCo-LDH對OER的影響,作者在飽和O2的1.0 M KOH溶液中評估了Rux SACs@FeCo-LDH的OER活性。如圖3(a)所示,在加入單原子Ru物種后,Ru1 SACs@FeCo-LDH表現出最好的OER活性,在10和500 mA cm2時,其超低過電位分別為194 mV和230 mV。同時,Ru1 SACs@FeCo-LDH的Tafel斜率最小,為25 mV dec-1,遠低于FeCo-LDH(48 mV dec-1)和Ru(93 mV dec-1)(圖3(b))。顯然,在較高的電流密度下,Ru1 SACs@FeCo-LDH仍然表現出良好的析氧動力學。圖3(c)顯示了在電流密度為1 A cm2時的過電位的比較。催化劑的OER活性增加的順序為:Ru <IrO2 <FeCo-LDH <RuO2 <Ru1 SACs@FeCo-LDH。Ru1 SACs@FeCo-LDH的最佳電催化OER活性甚至比之前報道的大多數催化劑都更好(圖3(d))。
此外,對于Ru1 SACs@FeCo-LDH,其在η=200 mV下的質量活性(MA)比FeCo-LDH高大約6倍,比Ru催化劑高大約2倍(圖3(e)),證實了由于Ru的引入,活性位點的增加。即使在更大的過電位下,它的MA達到了一個令人驚訝的值,遠遠超過了對比催化劑。此外,在η=200 mV處,周轉頻率(TOF) 計算結果為7.17X103 s-1,假設Co是Ru1 SACs@FeCo-LDH中的活性原子(圖3(f)),與報道的催化劑相比是最高的。
作者還利用電化學阻抗譜(EIS)證實了Ru1 SACs@FeCo-LDH的快速動力學(圖3(g))。結果發現,Ru1 SACs@FeCo-LDH的電荷轉移電阻(Rct)為在相同條件下,對照樣品中最小的。對于一種實用的電催化劑,在長期使用(超過1000小時)時保持良好的穩定性是很重要的,但這是非常具有挑戰性的,特別是在大電流密度下。如圖3(h)和(i)所示,Ru1 SACs@FeCo-LDH,在1 A cm2的電流密度下,在1200 h內僅降低了5%,甚至在20 000個連續循環后也只降低了10 mV,驗證了其顯著的長期穩定性。
同時,為了進一步推斷表面單原子對水分解動力學的作用,作者進行了HRTEM表征。如圖3(i)所示,經過OER過程后,HRTEM結果表明,沒有觀察到明顯的形態變化和原子分布。
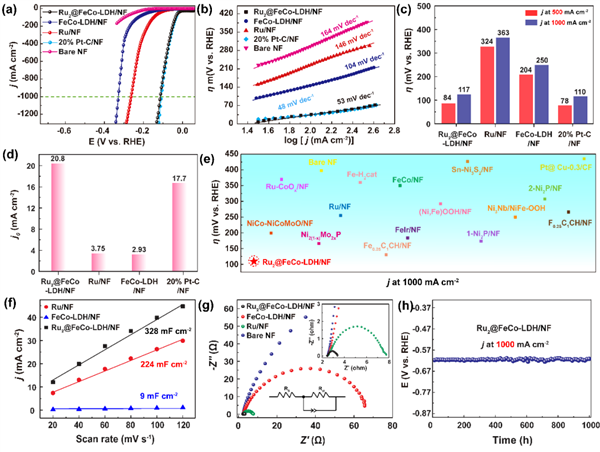
圖4 1M氫氧化鉀中的(a)極化曲線;(b)對應的Tafel圖;HER電催化在500 mA cm2和1 A cm2處的過電位的(c)比較;(d)交換電流密度的比較(j0);(e)在1.0 M氫氧化鉀溶液中,與最近報道的HER催化劑在1 A cm2的過電位相比;(f)計算的Cdl;(g)Nyquist圖;(h)Ru2 SACs@FeCo-LDH在維持1000 h所需電位的時間演化下的時間-電位曲線。@ The Authors
除OER外,作者還在堿性溶液(1.0 M KOH)中考察了不同樣品的HER電催化活性。如圖4(a)所示,增加Ru負載的Ru2 SACs@FeCo-LDH在接近零的起始過電位的堿性介質中表現出優越的HER活性,對應的Tafel斜率低至53 mV dec-1(圖4(b)),這與商用的Pt/C催化劑相似,且優于FeCo-LDH和Ru催化劑。如圖4(c)所示,為了達到500 mA cm2和1 A cm2的電流密度,Ru2 SACs@FeCo-LDH只需要84和117 mV的過電位,其堿性HER活性最好,與Pt/C非常接近。此外,與對比催化劑相比,Ru2 SACs@FeCo-LDH也表現出最大的交換電流密度(j0)值(圖4(d)),證實了其具有較高的反應性。值得一提的是,Ru2 SACs@FeCo-LDH的HER活性高于大多數報道的雜化電催化劑(圖4(e))。
同時,從圖4(f)圖中看到,Ru2 SACs@FeCo-LDH顯示最大的Cdl為328 mF cm2,和最小的Rct (圖4(g)), 表明更快的HER動力學是由于大大提高的電化學表面積和電荷轉移能力。重要的是,Ru2 SACs@FeCo-LDH顯示出良好的穩定性,在長期測試中降解可以忽略不計,在1000 h后在1 A cm2處的過電位損失可以忽略不計(圖4(h))。
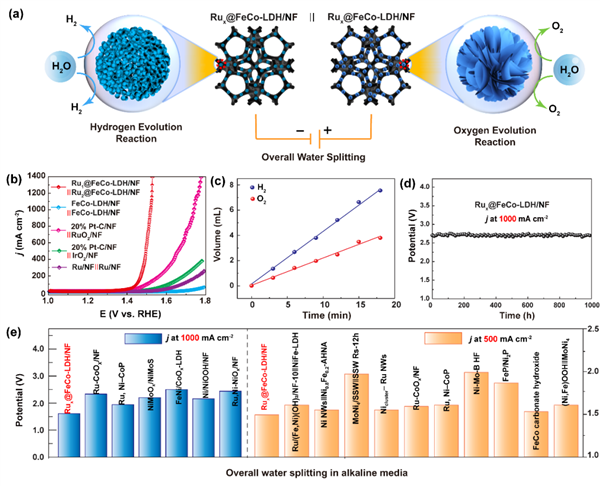
圖5 (a)整體水分解過程示意圖;(b)雙官能團Rux SACs@FeCo-LDH電催化劑的極化曲線;(c) Ru1 SACs@FeCo-LDH產生的氣體量與時間的關系,理論計算和實驗測量。Rux SACs@FeCo-LDH在相應的1 A cm2下1000 h的(d)時間-電位曲線;(e)報道的Rux SACs@FeCo-LDH在500 mA cm2和1 A cm2下的電池電壓與雙功能電催化劑的比較。@The Authors
考慮到Ru1 SACs@FeCo-LDH和Ru2 SACs@FeCo-LDH催化劑具有優異的OER和HER性能,作者分別組裝了一個以兩種催化劑為陽極和陰極的堿性電解槽(圖5(a))。令人印象深刻的是,在1.0 M KOH溶液中,這種電解槽在所有樣品中表現出最好的整體水分解活性(圖5(b))。此外,從圖5(c)可知,H2和O2的收集量約為2:1,法拉第效率接近100%,表明Rux SACs@FeCo-LDH對水分解具有較高的催化選擇性。更重要的是,在1 A cm2處連續測試超過1000小時,Rux SACs@FeCo-LDH的應用電位衰減可以忽略不計的,這表明其具有良好的耐久性(圖5(d))。所制備的Rux SACs@FeCo-LDH是一種很有前途的工業規模的整體水分解催化劑(圖5(e))。
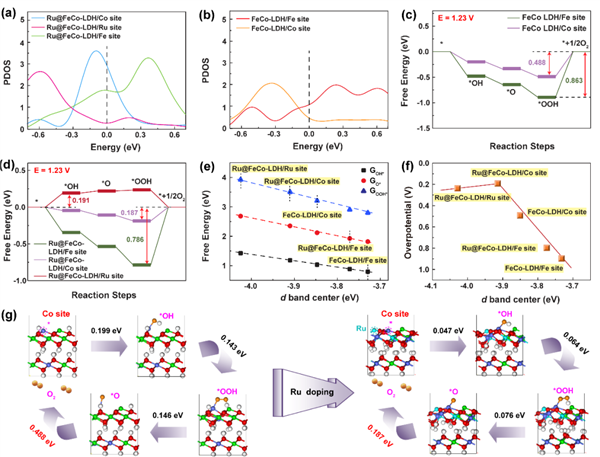
圖6 (a)和(b)計算了Ru SACs@FeCo-LDH和FeCo-LDH上的Fe、Co和Ru活性位點的部分態密度。(c)在FeCo-LDH上,和在 (d) Rux SACs@FeCo-LDH的OER過程的能量分布。(e)ΔGOOH*、ΔGOH*、ΔGO*與d帶中心之間的關系。(f)過電位作為d帶中心的函數的火山圖。(g)FeCo-LDH和Ru SACs@FeCo-LDH的Co活性位點上代表性OER機制。@The Authors
作者通過密度泛函理論(DFT)計算,闡明了堿性條件下摻雜在FeCo-LDH上的Ru物種(Ru SACs@FeCo-LDH)的電子結構的協同效應與催化活性之間的內在關系。如圖6(a)和(b)所示,Ru SACs@FeCo-LDH上的Co活性位點的d帶中心(0.098 eV)比FeCo-LDH(0.335 eV)更接近費米能級,表明在Ru SACs@FeCo-LDH在電化學反應中具有較強的給受電子能力。
相應地,堿性OER的反應途徑包括四個步驟(如圖6(c)和(d)所示),同時形成一系列中間體,如OH*、O*、OOH*、OO*。另外,作者根據活性位點的d軌道計算了d帶中心,得到了三種中間體(GOH*、GO*、GOOH*)的吸附自由能的變化曲線(圖6(e))。顯然,Ru原子的引入通過負位移的d帶中心優化了中間體的吸附自由能,從而促進了OER活性(圖6(e))。同時,如圖6(f)所示,當d帶中心適用時,OER的過電位可以達到“火山”峰值。通過比較FeCo-LDH,可以看到Ru單獨摻雜降低了過電位(圖6(f))。與FeCo-LDH和Ru SACs@FeCo-LDH界面上的Co位點相比,反應能量得到了顯著的優化(圖6(g))。因此,摻雜Ru原子降低了所有化合物的過電位,并擴大了活性催化位點。
05 成果啟示
本文獲得的Ru SACs@FeCo-LDH對于OER和整體水分解具有良好的電催化活性和穩定性,特別是在1000 mA cm2的大電流密度下。本文的策略為提高水分解工業規模制氫的單原子催化劑提供了新機遇。
06 參考文獻
Xueqin Mu, Xiangyao Gu, Shipeng Dai, et al. (2022), Breaking the symmetry of single-atom catalysts enables an extremely low energy barrier and high stability for large-current-density water splitting. Energy Environ. Sci., 2022, Advance Article.
https://doi.org/10.1039/D2EE01337A