EnergyChemNews 能源學人 2023-06-02 07:41 發(fā)表于廣東
近年來,電催化水分解過程中電極極化誘導的電極材料動態(tài)表面重構機制,以及如何利用表面重構技術實現(xiàn)催化活性的提升,已成為電催化領域重要的研究課題。因此,為促進電極材料表面重構工程的發(fā)展,本文系統(tǒng)評述了近年來過渡金屬基催化劑在電催化析氧反應(OER)和析氫反應(HER)中表面重構的研究現(xiàn)狀,包括: 1)表面重構的行為、起源及內外影響因素;2)表面重構衍生核殼異質結構的催化活性增益機制;3)基于預催化劑設計的重構調控策略;4)表面重構面臨的挑戰(zhàn)和機遇。本評述將為重構催化機制研究和高效電解水催化劑的設計和構筑提供科學的指導。
Surface reconstruction-derived heterostructures for electrochemical water splitting
Xu Luo, Xin Tan, Pengxia Ji, Lei Chen, Jun Yu, Shichun Mu*
EnergyChem, 2023, 5(2), 100091.
DOI: 10.1016/j.enchem.2022.100091
Graphical abstract
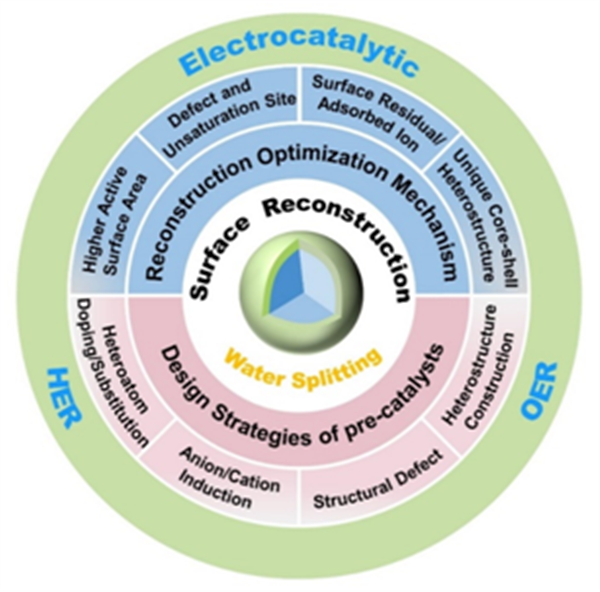
圖1. 基于預催化劑設計的電催化水分解表面重構優(yōu)化機理及調控策略。
全文鏈接:https://www.sciencedirect.com/science/article/pii/S2589778022000239
研究背景:
電催化水分解綠色制氫技術對可再生能源轉換與儲存具有重要意義。但要實現(xiàn)電催化水分解制氫技術的廣泛應用,開發(fā)高效的電催化水分解催化劑,降低析氫(HER)和析氧(OER)兩個半反應能壘是關鍵。當前,隨著各種過渡金屬基催化劑蓬勃發(fā)展,其在電催化OER和HER過程中的動態(tài)結構重構也成為研究的焦點(Figure 2)。由于大多數過渡金屬化合物在電化學OER條件下熱力學不穩(wěn)定,它們傾向于通過動態(tài)結構演化達到相對穩(wěn)定的狀態(tài)。由此原位衍生的重構表面則作為真正的反應活性物質將引起催化活性的變化,為催化機制的研究帶來挑戰(zhàn),同時也使利用表面重構設計新穎的異質結構催化劑成為可能。
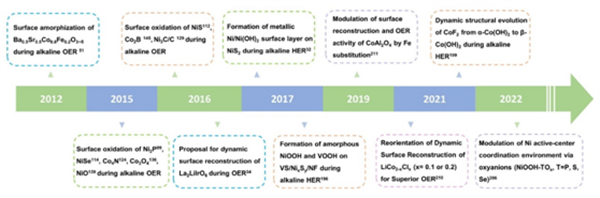
圖2. 電催化水分解催化劑重構工程發(fā)展的時間軸。
內容簡介:
在這篇評述中,作者重點介紹了電催化OER和HER過程中的表面重構。
1. 表面重構
在標準條件下,HER和OER對可逆氫電極(RHE)的熱力學平衡勢(E0)分別為0 V和1.23 V。由于反應動力學緩慢,且電解槽內存在電阻,為了驅動分子轉化催化,實際電催化中需要施加比平衡電位更大的負電位或正電位。當施加電位超過預催化劑中金屬位點的氧化還原電位時,可能導致預催化劑表面結構發(fā)生重大變化,并產生新的物種,導致核殼異質結構形成或完全轉化為重構相。這也被分別定義為表面重構和完全重構。催化劑的重構動力學及表面重構程度受外在因素(電解液pH、施加電位、測試溫度等)和內在因素(預催化劑的結構)的雙重影響。
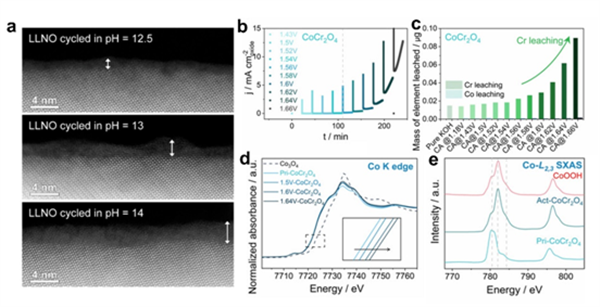
圖3.(a)不同pH的KOH電解液中循環(huán)的La2Li0.5Ni0.5O4 的HAADF-STEM圖像。(b)CoCr2O4在不同電位(1.43 ~ 1.66 V)下的連續(xù)計時電流(CA)曲線。(c)CoCr2O4每隔20分鐘進行一系列CA測試后的元素浸出累積量。(d)原始CoCr2O4和在1.5/1.6/1.64 V電位下CA測試 7小時后活化CoCr2O4的歸一化Co k邊XANES光譜。(e) Pri-CoCr2O4和Act-CoCr2O4的Co L邊軟XAS光譜。
2. 電催化OER/HER過程中的表面重構
基于電催化OER和HER過程中的表面重構,作者分別介紹了OER和HER的催化機理,并評述了一些代表性的表面重構衍生異質結構析氫/析氧催化劑。在電催化OER過程中,大多數催化劑,特別是非貴金屬催化劑,通常會發(fā)生表面重構,并原位形成低結晶度的氧化物或羥基氧化物。目前,過渡金屬基硫族化物、磷化物、氮化物、碳化物、硼化物、氟化物、氧化物、氫氧化物及合金類預催化劑在OER電催化過程中的表面重構已被廣泛報道。與OER電催化中氧化重構的廣泛研究相比,關于HER過程中催化劑結構演變的報道相對較少。大多數研究聲稱在HER催化過程中催化劑的組成、化學狀態(tài)和形態(tài)沒有明顯變化。然而,最近的一些研究卻揭示了催化劑在HER陰極電位驅動下的氧化/氫氧化或還原行為。
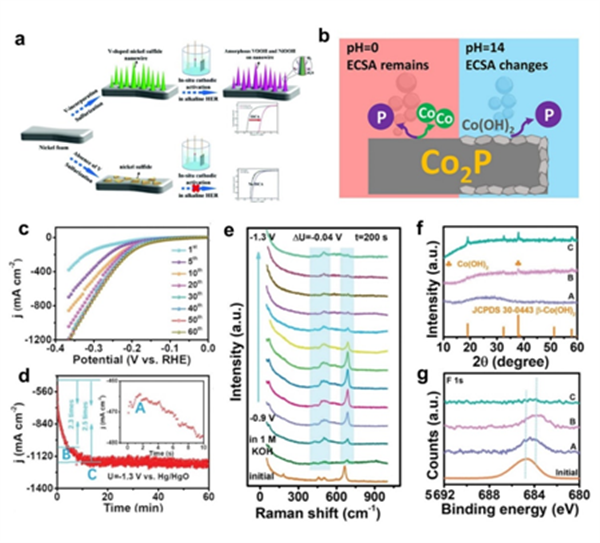
圖4.(a)VS/NixSy/NF的原位陰極活化示意圖。(b)Co2P@CP在酸性(pH = 0)和堿性(pH = 14) HER條件下的表面重構示意圖。CoF2的(c)連續(xù)LSV曲線和(d) i-t曲線。(e) CoF2在堿性HER中的原位拉曼光譜。(f)CoF2在不同i-t曲線時間點的XRD圖譜和(g) F1s高分辨率XPS光譜。
3. 表面重構優(yōu)化機制
目前報道的預催化劑重構大多伴隨著催化活性的提高。因此,電催化重構也被認為是催化劑的一種結構自優(yōu)化行為。此外,實驗表明,重構衍生的活性物質通常比直接合成的活性物質具有更高的催化活性,同源金屬化合物衍生的同源重構層也普遍表現(xiàn)出不同的催化活性。深入解析表面重構優(yōu)化機制,識別重構活性表面的構效關系,對預催化劑的設計和構筑具有重要意義。在此部分內容中,作者深入分析了一些可能的表面重構優(yōu)化機制,如增加的活性表面積、更多的缺陷和不飽和位點形成、重構表面殘余/吸附離子修飾及獨特的重構衍生核殼異質結構等,以幫助了解重構層的活性起源和同源重構層的催化活性差異。
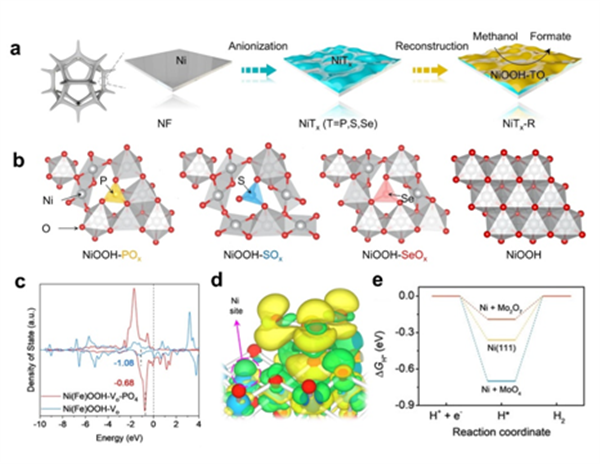
圖5.(a)NiOOH-SOx(T = P, S, and Se)電催化劑的合成示意圖。(b)不同氧陰離子配位的NiOOH和純NiOOH的優(yōu)化結構模型。(c) Ni(Fe)OOH-VO和Ni(Fe)OOH-VO-PO4Ni 3d軌道的DOS。(d) Ni(Fe)OOH-VO-PO4的差分電荷密度。(e) Ni(111)和吸附MoO4和Mo2O7的Ni(111)上HER的自由能圖。
4.預催化劑設計策略
電催化過程中的結構重構對催化活性的調節(jié)起著重要的作用,如何利用結構重構構建出具有優(yōu)異活性和穩(wěn)定性的電催化劑也成為電催化領域的研究熱點。因此,作者重點討論了調控結構重構的一些預催化劑的設計策略,包括雜原子摻雜/取代、陰離子/陽離子誘導、結構缺陷、異質結構構建等。
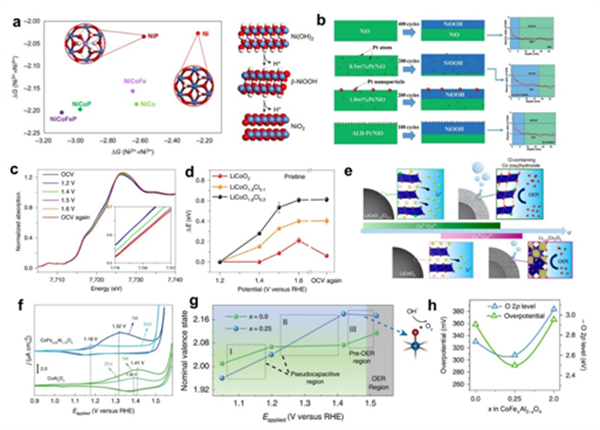
圖. 6.(a)DFT計算的不同Ni位點取代的吉布斯自由能變化(Ni2+→Ni3+→Ni4+)。(b)500次循環(huán)后催化劑的相重構行為及相應的XPS深度分布圖。(c)LiCoO1.8Cl0.2在不同電位下的Co K-edge XANES光譜。(d)LiCoO2?xClx(x= 0.1 or 0.2) 和LiCoO2在不同電位下的Co K-edge偏移。(e)LiCoO1.8Cl0.2 and LiCoO2在OER過程中的原位表面重構示意圖。(f)CoAl2O4和CoFe0.25Al1.75O4在CV循環(huán)過程中的贗電容行為。(g)CoAl2O4和CoFe0.25Al1.75O4在1.05, 1.20, 1.42 和1.52?V versus RHE電位下Co的氧化態(tài)。(h)CoFe0.25Al1.75O4 (x=?0.0, 0.25 和2.0)在10?μA?cm-2ox處的OER過電位和相對于費米能級的O2p帶中心。
雖然現(xiàn)階段預催化劑的表面重構研究已經取得了一定的進展,但問題和挑戰(zhàn)仍然存在,需要作更深入的探索:
1. 準確識別表面重構層的配位結構是揭示催化機理的關鍵。因此,應用表面敏感的原位表征技術來實時捕獲催化劑近表層的結構信息更具說服力。此外,重構層與預催化劑的協(xié)同作用不可忽視。
2. 前仍然缺乏對重構殼層厚度、界面效應以及原始預催化劑內核在表面重構異質結構中作用的系統(tǒng)研究。
3. 對重構體系本征催化活性的評價有待進一步完善。電催化重構過程也會相應,導致表面粗糙化和電化學活性面積(ECSA)的動態(tài)變化,為了研究催化活性的真正來源和重構引起的本征活性的動態(tài)變化,需要提供ECSA歸一化的電流密度。此外,對ECSA的不適當估計也會誤導對本征活性的分析。
4. 電化學測試方法的差異性為系統(tǒng)性的比較分析帶來困難。目前,用于研究重構的電化學方法主要有循環(huán)伏安(CV)、計時電位和計時電流分析方法。然而,這些電化學測試方法與重構之間的聯(lián)系還沒有得到深入的研究,對于重構的評價缺乏分析標準。因此,未來應致力于在重構工程領域建立統(tǒng)一的標準,以幫助分析和關聯(lián)不同研究工作中的重構信息,從而進一步解析預催化劑的重構機理。
5. HER和其他電化學反應(氧還原反應、CO2還原反應等)過程中涉及的結構重構還有待進一步研究。此外,陰極電位誘導的還原表面在含氧條件下容易被氧化,在一定程度上會影響對重構行為的準確識別。
作者簡介:

羅旭 武漢理工大學在讀博士
主要研究方向為OER/HER非貴金屬催化劑的設計與構建及其在電催化反應中的重構行為。