
�_�h(hu��n)�ۺ���һ�(xi��ng)�ϳ���朹��ܸ߷��Ӳ��ϵ���Ҫ���ԡ�����C(sp3)�CN�I�V���������S��h(hu��n)���ЙC(j��)�������У�������Ч���Ѵ���W(xu��)�I�������_�h(hu��n)�ۺ��ṩһ�N�·f��;����Ȼ����C(sp3)�CN�I�����^�ߵ��I���x�ܣ���(d��o)��������y���ஔ(d��ng)����Ŀǰ���о���Ҫ��ه�ڸ߭h(hu��n)�������w���絪��ड����s�h(hu��n)�����Ť�����������(q��)��(d��ng)C(sp3)�CN�I�������Ԍ�(sh��)�F(xi��n)�_�h(hu��n)�ۺϡ����ڟo���@�h(hu��n)�����ĭh(hu��n)����w�Ќ�(sh��)�F(xi��n)C(sp3)�CN�I�����ѣ���Ȼ���R����������(zh��n)���D1����
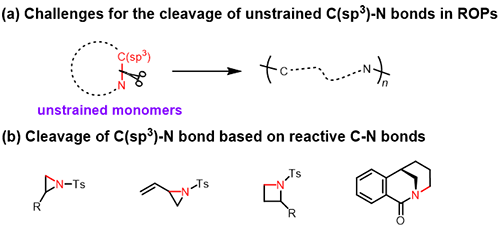
�D1. C(sp3)-N�I�������_�h(hu��n)�ۺ��еđ�(y��ng)��
��ɽ��W(xu��)�S�h���n�}�M��Ҫ�����������ɻ��ۺϼ���h(hu��n)�_�h(hu��n)�ۺϷ������о���ּ�ڞ���朹��ܸ߷��Ӳ��ϵĿɿ��Ƃ��ṩ��Ч�ĺϳɼ��g(sh��)����ǰ�ڹ����У������״��������~�ˠ��ӳ�-�������C(j��)�Ƴɹ���(sh��)�F(xi��n)�˟o�����h(hu��n)����w��C(sp3)�CS�I���x�����Д࣬��(g��u)����һ��·f���_�h(hu��n)�ۺϷ���(y��ng)�wϵ�����ɹ��Ƃ��˷������ɿء��ֲ�խ���܉������Ƕ��������F(tu��n)�ĺ���߷��Ӳ�����Angew. Chem. Int. Ed. 2023, 62, e202217895�����ڱ��(xi��ng)�о��У������c�A��������W(xu��)�ܵ��Ž����n�}�M�������M(j��n)һ����ԓ������չ���o�����h(hu��n)����w�е�C(sp3)�CN�I���ѣ��״Ό�(sh��)�F(xi��n)�˻����~�ˠ��ӳ�-�����C(j��)����C(sp3)�CN�I�����_�h(hu��n)�ۺϷ���(y��ng)���D2����ͬ�r(sh��)���ęC(j��)���Ƕȳ��l(f��)���������������l(f��)չ���@��ۺϷ���(y��ng)��ʽ���������~�ˠ��ӳ��C�����_�h(hu��n)�ۺϣ�Michael Addition�CFragmentation Ring�COpening Polymerization, MAFROP, /![]() �������Ա����ϵ�y(t��ng)���_�l(f��)���о��@��������~�ˠ��ӳ�-�������C(j��)�Ƶ��_�h(hu��n)�ۺϷ���(y��ng)��
�������Ա����ϵ�y(t��ng)���_�l(f��)���о��@��������~�ˠ��ӳ�-�������C(j��)�Ƶ��_�h(hu��n)�ۺϷ���(y��ng)��
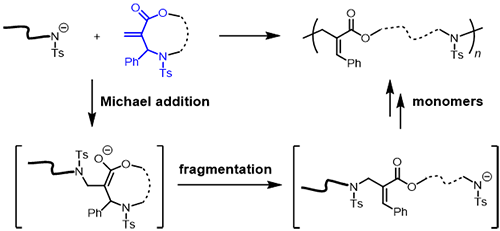
�D2. ����C(sp3)-N�I���ѵ��~�ˠ��ӳ�-�����_�h(hu��n)�ۺϷ���(y��ng)
�ڱ��(xi��ng)�о��У����������O(sh��)Ӌ(j��)���ϳ������ͭh(hu��n)����wM1�������Ҝؿ՚�l���ºY�x�˲�ͬ�����l(f��)�����Y(ji��)��������N-�һ���������⛣�Init2��չ�F(xi��n)����(y��u)���ľۺϿ������ܣ����þۺ������Փ��������Mn,theo���c�˴ŷ�������Mn,NMR���߶��Ǻ����D3�������(y��u)����(y��ng)�l���£����в�̼ͬ��L(zh��ng)�ȵĆ��wM1�CM3���܌�(sh��)�F(xi��n)�ɿؾۺϡ�ͨ�^�{(di��o)�؆��w�c���l(f��)���ı������ɹ��ϳ��˷��������{(di��o)���ֲ�խ�ľۺ��ֵ��һ����ǣ����þۺ�������������Y(ji��)��(g��u)���ʽ��(g��u)�͞�����>90% cis�������⣬ͨ�^�{(di��o)��(ji��)�h(hu��n)�ߴ磬�����`��ؿ�������е�ԭ�ӵ��g�࣬�@��(du��)�ڂ��y(t��ng)���w���絪��ऺ͵��s�h(hu��n)���飩���ԘO������(zh��n)�ԡ�
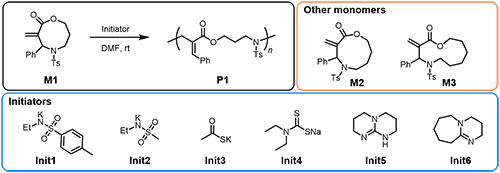
�D3. �ۺϷ���(y��ng)�о�
��(d��ng)���W(xu��)�о�������ԓ�ۺϷ���(y��ng)��ѭһ��(j��)����(y��ng)��(d��ng)���W(xu��)Ҏ(gu��)�ɣ���(sh��)����������Mn���c�D(zhu��n)���ʳʾ����P(gu��n)ϵ��������(g��)�ۺ��^���з������ֲ������^խˮƽ��![]() < 1.1�����M(j��n)һ����MALDI-TOF�|(zh��)�V�����@ʾ��ÿ�M�����g���Ĕ�(sh��)ֵ�;ۺ����؏�(f��)��Ԫ�ķ�������ͬ���Ҿۺ������������ĩ�˽Y(ji��)��(g��u)�����ڴˣ������M(j��n)��������쌍(sh��)�(y��n)���ɹ��Ƃ��˲�ͬ��Ƕ�ι�������ȣ�ͨ�^M1�ľ��ۺϳɴ������D(zhu��n)�Ƅ�P1���S��һ偷�����M2�M(j��n)������죬�ɹ��õ��˶�Ƕ�ι�����P1-b-P2��ͬ����ʹ�Æ��wM1��M3����ɹ��Ƃ�Ƕ�ι�����P1-b-P3�����⣬ͨ�^������M1��M2��M3�܉�һ偷��Ƃ���Ƕ�ι�����P1-b-P2-b-P3�����܉�(zh��n)�����ۺ��������������ֲ����@Щ��(sh��)�(y��n)���H�(y��n)�C��MAFROP�Ŀɿ��ԣ���ͻ����ԓ�����ںϳɾ������_�Y(ji��)��(g��u)�ۺ��﷽��ĸ�Ч�ԡ�
< 1.1�����M(j��n)һ����MALDI-TOF�|(zh��)�V�����@ʾ��ÿ�M�����g���Ĕ�(sh��)ֵ�;ۺ����؏�(f��)��Ԫ�ķ�������ͬ���Ҿۺ������������ĩ�˽Y(ji��)��(g��u)�����ڴˣ������M(j��n)��������쌍(sh��)�(y��n)���ɹ��Ƃ��˲�ͬ��Ƕ�ι�������ȣ�ͨ�^M1�ľ��ۺϳɴ������D(zhu��n)�Ƅ�P1���S��һ偷�����M2�M(j��n)������죬�ɹ��õ��˶�Ƕ�ι�����P1-b-P2��ͬ����ʹ�Æ��wM1��M3����ɹ��Ƃ�Ƕ�ι�����P1-b-P3�����⣬ͨ�^������M1��M2��M3�܉�һ偷��Ƃ���Ƕ�ι�����P1-b-P2-b-P3�����܉�(zh��n)�����ۺ��������������ֲ����@Щ��(sh��)�(y��n)���H�(y��n)�C��MAFROP�Ŀɿ��ԣ���ͻ����ԓ�����ںϳɾ������_�Y(ji��)��(g��u)�ۺ��﷽��ĸ�Ч�ԡ�
������������ԓ�ۺϷ���(y��ng)���C(j��)���c���w�x���ԁ�Դ�������M(j��n)�����ܶȷ�����Փ��DFT��Ӌ(j��)�㡣�Y(ji��)���������~�ˠ��ӳ��܉�������-�����܉����������ߞ�Q�ٲ�������(d��o)�a(ch��n)������w��(g��u)�͡����������ߌ�(du��)��-�����^�����γ�혷��a(ch��n)��ăɷN�����^�ɑB(t��i)��cis-TS2�ctrans-TS2���M(j��n)����Ӌ(j��)�㡣Ӌ(j��)��Y(ji��)���@ʾ���^�ɑB(t��i)cis-TS2��14.8 kcal/mol�����܉��@������trans-TS2���܉���25.5 kcal/mol�����f���ʽ����������γɸ��������M(j��n)һ�����^�ɑB(t��i)�Y(ji��)��(g��u)�����l(f��)�F(xi��n)����Ԫ�h(hu��n)��(n��i)Et(Ts)NCH2-���F(tu��n)�c��ԭ��֮�g�Ŀ�h(hu��n)����������trans-TS2��������cis-TS2��ԭ�����D4���������(y��n)�C�@һ�Ɯy(c��)�����ߺϳ��˷ǭh(hu��n)����ﲢ�M(j��n)���ˌ�(du��)�Ȍ�(sh��)�(y��n)���Y(ji��)���������a(ch��n)����Ҫ�鷴ʽ��(g��u)�ͣ��M(j��n)һ���C��(sh��)�˭h(hu��n)����w�_�h(hu��n)�ۺ����^�쵽���^���ʽ���w�x������ҪԴ�ڿ�h(hu��n)�������(q��)��(d��ng)��
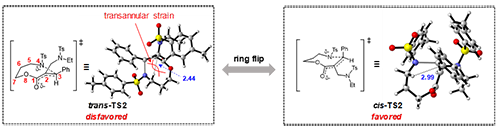
�D4. �ۺϷ���(y��ng)�C(j��)�����о�
���⣬�����M(j��n)һ���� MAFROP ������չ���c N-������������Ĺ��������D5����ͨ�^��׃����w��Ͷ����ɹ��Ƃ��˃ɷNǶ�ι�����P1-b-PTsMAz��PTsMAz-b-P1����ͨ�^DOSY�V�C��(sh��)��Ƕ�ι�����ijɹ��ϳɡ��S�������ü״��c̎����Ƕ�ι�����PTsMAz-b-P1�r(sh��)��P1�οɱ��x���Խ��⣬��PTsMAz�α��ַ�(w��n)����չʾ��ԓ�����ھۺ��﹦�ܻ����������{(di��o)�ط���đ�(y��ng)�Ý�����
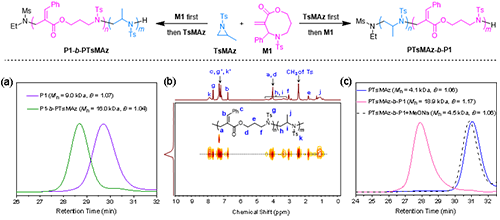
�D5. �cN-����������वĹ����о�
������߲���ģ�K���������������c�_�h(hu��n)�C(j��)�P(gu��n)�Y(ji��)�ϣ���(ji��n)����ϳ�������ABCDE��Ԫ���еĴ�h(hu��n)���wM4��ͨ�^��(du��)M4�M(j��n)���_�h(hu��n)�ۺϣ��ɹ���(g��u)���˷������ɿ��ҷֲ�խ��������пɿؾۺ������D6����ֵ��ע����ǣ��@ۺ���ͨ���y��ͨ�^�ۺϣ���ȱ�c(di��n)���ڷ��������ɿأ�����y(t��ng)�_�h(hu��n)�ۺϣ���ȱ�c(di��n)�����y�����������������ָ���Ĺ��܈F(tu��n)���ȷ����M(j��n)���Ƃ䡣�@һ�Y(ji��)����(bi��o)־��MAFROP��������朹��ܸ߷��Ӳ����ɿ��Ƃ������~�����P(gu��n)�Iһ����
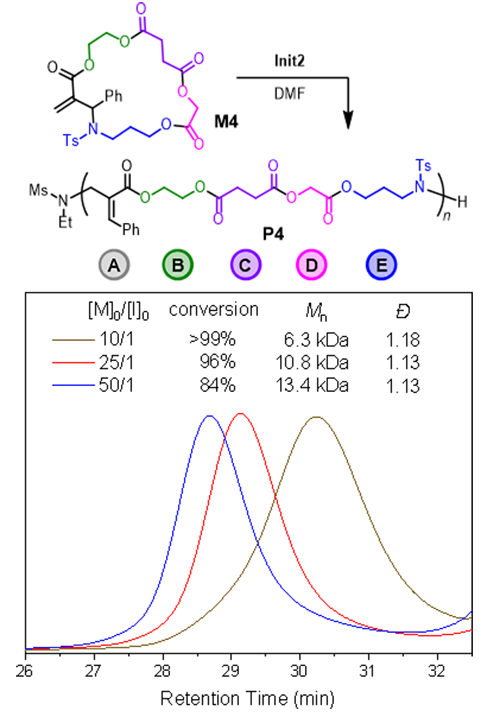
�D6. ���пɿؾۺ���ĺϳ�
���Y(ji��)�����о���(chu��ng)���Ե��_�l(f��)��һ�N����C(sp3)�CN�I���ѵ��~�ˠ��ӳ��C�����_�h(hu��n)�ۺϣ�MAFROP���·���(y��ng)�wϵ��ԓ�ۺϷ���(y��ng)���ڜغ͗l���¸�Ч�M(j��n)�У��ϳɷ������ɿء��ֲ�խ�Ҿ����ʽ���w�x���Ե��������۰������⣬ԓ�ۺϷ����M(j��n)һ���U(ku��)չ���cN-����������वĹ����Լ����пɿؾۺ���ĺϳɣ�չʾ��ԓۺϷ���(y��ng)���Ƃ���ж��ӻ���朽Y(ji��)��(g��u)�Ĺ��ܸ߷��Ӳ��Ϸ���ďV韑�(y��ng)��ǰ����ԓ�о���������Aza-Michael Addition�CFragmentation Ring�COpening Polymerization�����}�l(f��)�������������W(xu��)��(hu��)��(hu��)־����J. Am. Chem. Soc. 2025, DOI: 10.1021/jacs.5c03181������һ���ߞ��S��ͬ�W(xu��)�����x������Ȼ�ƌW(xu��)����͏V�|ʡ��Ȼ�ƌW(xu��)������(du��)���(xi��ng)Ŀ���Y����ͬ�r(sh��)Ҳ���x���о��^���нo����������ѡ��ώ��͌W(xu��)����
ԭ��朽ӣ�https://pubs.acs.org/doi/10.1021/jacs.5c03181
- ��ƴ����F(tu��n)�(du��) Angew���Y(ji��)�� / �����(q��)��(d��ng)��-������(n��i)����ƽ���_�h(hu��n)�ۺ��Ƃ���Ѓ�(y��u)��������ܵ��]��ѭ�h(hu��n)���������� 2025-05-14
- �Ĵ���W(xu��)�섦�����ڈF(tu��n)�(du��) Angew���_�h(hu��n)�ۺ��Ƃ仯�W(xu��)�ɻ��յĸ����ܰ뷼���Ծ����� 2025-04-11
- �����၆������W(xu��)ͬ�V���ڈF(tu��n)�(du��) Angew: ���ڿɿ��_�h(hu��n)�ۺϵĸ߷����������wҎ(gu��)���ĭh(hu��n)��ܻ��ۣ���-�u���ᣩ 2025-04-09
�\���P(gu��n)ע�߷��ӿƼ�

- ���ܹ��A��朆���������22��...
- ����(b��o)���C(j��)��(hu��)��500+ˎ��...
- ���HƷ�ơ�ǰ�ؕ�(hu��)�h�cչλ�D...
- ������AI�ǿء���Ч�����x��...
- 2025�ؑc����չ���c(di��n)������...
- ���a(ch��n)������٣�������^�Ƽ�...
- 2025Ϳ��ԭ���x�ϴ��(hu��)
- �Ј�(ch��ng)��300�|��PMEC China ɫ...
- ����ע��(c��)��2025����������չ...
- ���a���υ��^�A(y��)��ӛ���öY��...
- �ۺ���l(f��)���ИI(y��)���g(sh��)��Ӗ(x��n)����...
- �����츣��/��혡�������ǻ...
- ��ɽ��W(xu��)�Ƕ�ؔ(c��i)/�S�ſ�/����...
- �Ї��r(n��ng)�I(y��)�ƌW(xu��)Ժ����о�����...
- ��������� Adv. Sci.: �ň�(ch��ng)...
- ���t(y��)���F(tu��n)�(du��)���������R��...
- ������/���ƴ�Ժ����ƽ...
- �B�T��W(xu��)�܌W(xu��)������ Macromo...
- ɽ���܂������ڡ���(j��)���w�ɷ�...
- �A���r(n��ng)������p���ڡ�����x...
- �Ї��ƴ���Ƙ�/���������...
- �B�T��W(xu��)������ڈF(tu��n)�(du��) AFM��...