自從單原子催化劑(SAC)被發現以來,其催化活性來源的研究一直是中心科學問題。富含雜原子的碳基SAC由于其良好的導電性、高比表面積以及與金屬原子良好的相容性,成為所有SAC中被研究最多的一類催化劑材料。這種良好的相容性被廣泛認為來源于碳和雜原子的強配位能力。在早期的研究中,具有氮配位單金屬位點(M-N4)的碳材料是最受歡迎的SAC。不久之后,研究人員認識到碳基SAC中的配位環境應該是N-/C-共配位的單金屬位點 (M-NxC4-x),而不是單一的M-N4,但是僅通過X射線吸收光譜(XAS)和相應的擬合技術無法區分C和N(圖 1)。只能勉強根據密度泛函理論計算和催化性能構建一定程度的構效關系理解,一些M–NxC4–x位點比相應的M–N4位點表現出更高的催化活性,或者甚至相反的結論。

最近,上海交通大學莊小東教授課題組以N-混雜卟啉金屬配合物(NCPor-Ms)和傳統卟啉金屬配合物(Por-Ms)為研究對象,分別研究了M-N3C1和M-N4位點的本征電催化氧還原活性。該工作通過理論計算預測了21種不同金屬中心的NCPor-Ms和Por-Ms的催化活性,并分別建立了M-N3C1和M-N4位點的ORR活性火山曲線(圖1)。其中,位于火山曲線頂點的NCPor-Co在NCPor-Ms中表現出最高的催化活性,理論起始電位為 0.77 V。為了驗證 M-N3C1和M-N4位點的理論結果,作者合成了6種不同金屬中心的NCPor-Ms和Por-Ms(M = Mn、Co、Ni、Cu、Pd 和 Ag),并通過擴展 X 射線吸收精細結構(EXAFS)證實了M-N3C1位點的配位環境。根據電化學測量,NCPor-Co表現出最高的半波電位(0.83 V vs. RHE;0.1 M KOH),且遠高于Por-Co(0.77 V vs. RHE,圖2)。原位XAS表明Co-N3C1位點在催化ORR過程中會形成Co(III)物種(圖3)。此外,作者通過原位衰減全反射表面增強紅外吸收光譜(ATR-SEIRAS)監測到了反應中間體(*O2、*OOH)。密度泛函計算表明,NCPor-Co的高ORR活性歸因于其較低的帶隙和最佳的d帶中心(圖4)。這項工作為不對稱單金屬位點的本征催化活性研究提供了全面性的理解。
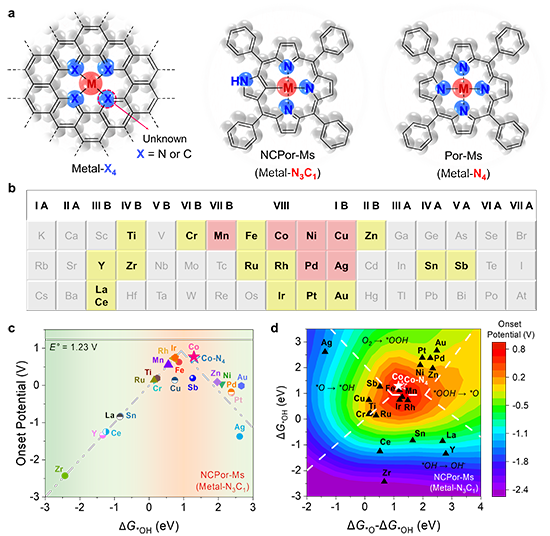
圖1. M-N3C1位點的理論活性。(a) 碳基單原子催化劑、NCPor-Ms、Por-Ms的結構示意圖;(b) 本工作中研究的21種金屬元素。理論計算研究(黃色);理論計算和實驗驗證(粉色);(c) M-N3C1位點的氧還原火山圖曲線;(d) M-N3C1位點的催化活性等高線圖。
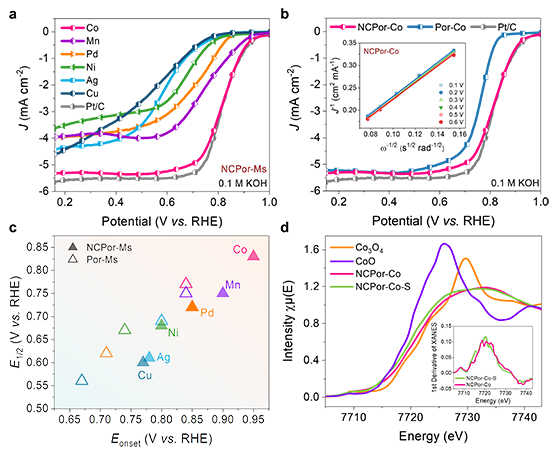
圖2. NCPor-Ms氧還原催化活性研究。(a) NCPor-Ms在氧氣飽和的0.1 M KOH溶液中的電化學極化曲線;(b) NCPor-Co和Por-Co的電化學極化曲線;(c) NCPor-Ms和Por-Ms的起始電位和半波電位的總結圖;(d) CoO、Co3O4、NCPor-Co及其穩定性測試后的XANES譜圖。插圖:XANES譜圖的一階導數。
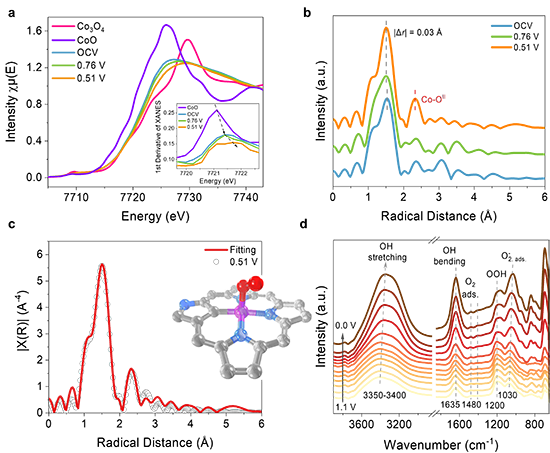
圖3. Co-N3C1位點電催化ORR的原位表征。(a) CoO、Co3O4和NCPor-Co(不同電壓下:OCV、0.76 V、0.51 V vs. RHE)的原位XANES譜圖。(b) NCPor-Co的原位EXAFS譜圖;(c) O2吸附的NCPor-Co的EXAFS擬合曲線;(d) NCPor-Co的原位ATR-SEIRAS譜圖。
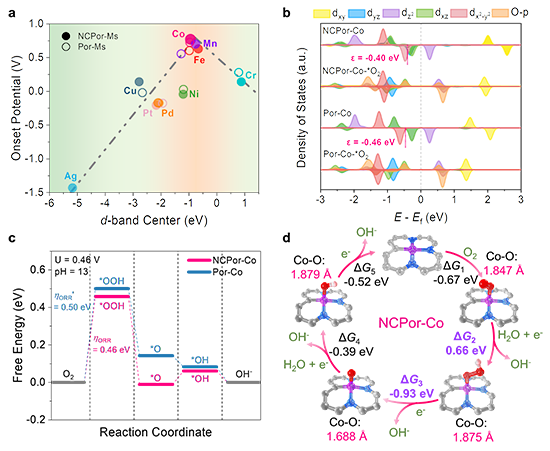
圖4. Co-N3C1的催化機理。(a) M-N3C1和M-N4活性位點的理論d帶中心; (b) NCPor-Co、Por-Co、O2吸附的NCPor-Co和O2吸附的Por-Co的態密度圖。(c) Co-N3C1和Co-N4位點的催化ORR自由能能壘圖;(d) 堿性介質中Co-N3C1位點的氧還原循環反應圖。
目前該工作以“Well-defined N3C1-anchored Single-Metal-Sites for Oxygen Reduction Reaction”為題在線發表在《德國應用化學》(Angewandte Chemie International Edition, 2023, DOI: 10.1002/anie.202314833)上。該論文的第一作者為上海交通大學2021級博士研究生黃森鶴。該項工作得到了基金委面上、優秀青年基金、科技部重點研發、上海市科委等經費資助。
原文鏈接:https://doi.org/10.1002/anie.202314833
